Terapia
Al momento, la terapia della Sindrome di Dravet (SD) è sintomatica cioè mira a controllare le crisi epilettiche. Purtroppo, in quasi tutti i pazienti, le crisi sono farmacoresistenti e tendono a persistere anche nell’età adulta.
La riduzione della frequenza delle crisi è un obiettivo importante da raggiungere in quanto può garantire una migliore qualità di vita ed un potenziamento delle performances quotidiane, consentendo così al bambino prima, e poi all’adulto, di compiere progressi e di acquisire una maggiore autonomia nella vita quotidiana.
Durante la prima e la seconda infanzia può essere utile evitare specifici fattori che possono favorire le crisi, come ad esempio impedire rapidi cambiamenti di temperatura corporea o ridurre al minimo le stimolazioni fotiche e visive.
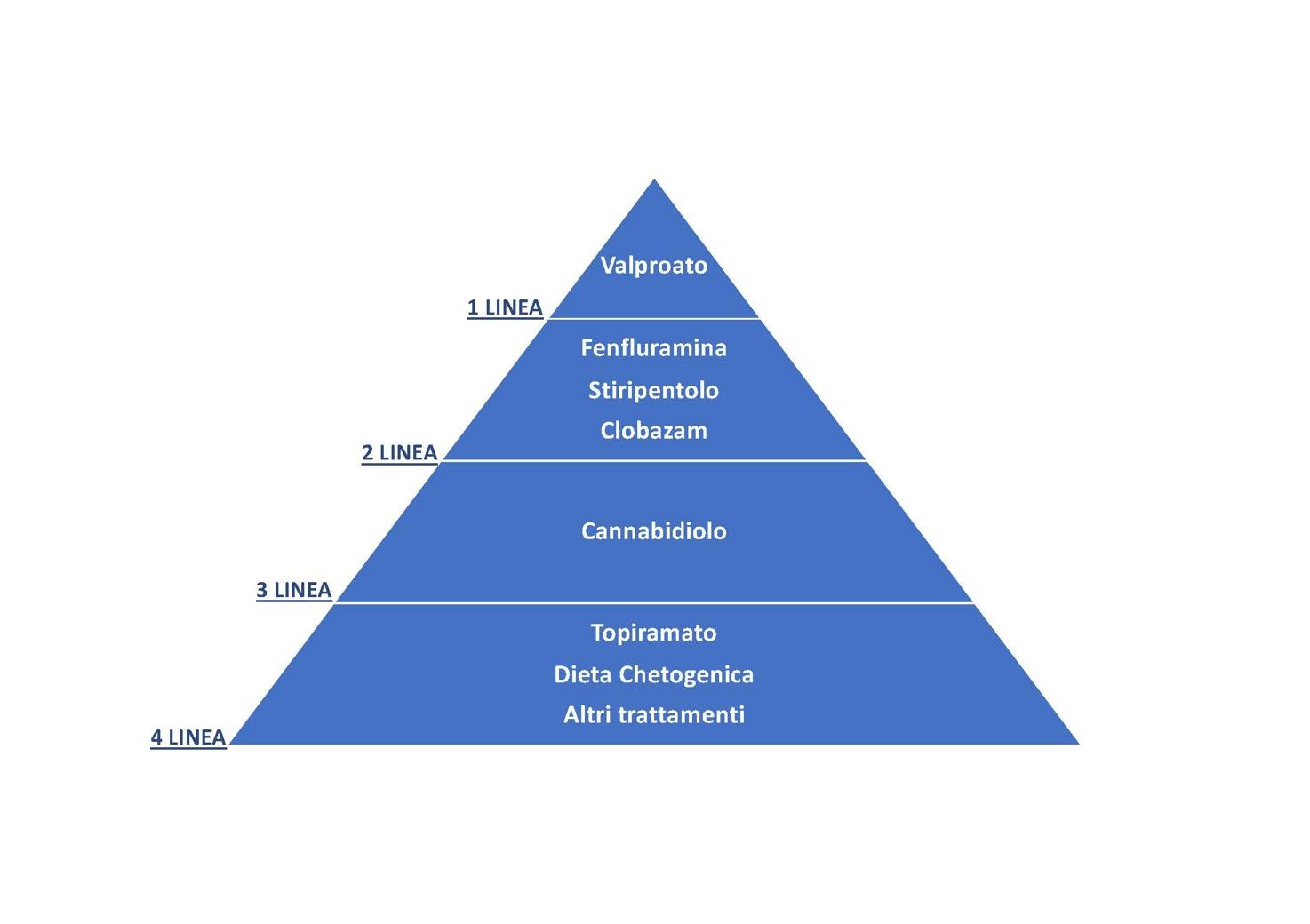
Spesso i pazienti assumono una politerapia farmacologica, facendo uso di più farmaci in combinazione tra di loro. I farmaci raccomandati per la SD sono riepilogati nella figura che segue, con evidenza dei farmaci di prima, seconda, terza e quarta linea in base a criteri definiti da clinici esperti e secondo lo schema proposto sulla base di un recente Consenso Internazionale.
Va tenuto presente che, nel sospetto di SD, anche prima dell’analisi genetica, è raccomandato evitare l’utilizzo di farmaci sodio-bloccanti, tra cui carbamazepina, oxcarbazepina, eslicarbazepina, lacosamide, lamotrigina, fenitoina, così come sono sconsigliati tiagabina, vigabatrin, gabapentin e pregabalin. Nei pazienti Dravet questi farmaci possono infatti peggiorare la sintomatologia epilettica.
È importante sottolineare che le persone affette da SD possono presentare un’ampia gamma di manifestazioni, più o meno gravi, e diversi tipi di crisi, i pazienti rispondono alla terapia in modo
diverso l’uno dall’altro. La scelta della terapia migliore varia da paziente a paziente in base all’età, alla storia clinica e ad altri fattori specifici, e perciò deve essere effettuata solo con il proprio medico curante, che conosce la storia del paziente e le sue caratteristiche, sulla base anche delle aspettative del paziente e dei familiari.
Purtroppo, la maggior parte dei pazienti non potrà raggiungere la completa libertà dalle crisi, ma massimizzare la qualità della vita e limitare gli effetti collaterali dei farmaci dovrebbero essere delle priorità tanto quanto il controllo delle crisi stesse.
Il valproato, o acido valproico, (Depakin) è considerato il farmaco di prima linea nel trattamento della SD.
Anche la fenfluramina (Fintepla) e lo stiripentolo (Diacomit) si sono dimostrati efficaci, così come il farmaco di terza linea, il cannabidiolo (Epidiolex). Il clobazam, utilizzato in aggiunta agli altri principi attivi, è un farmaco sicuro, ben tollerato e con attività ad ampio spettro su tutti i tipi di crisi.
La fenfluramina è stata approvata come terapia aggiuntiva per il trattamento di crisi associate alla SD a partire dai due anni di età. È importante ricordare che la somministrazione di fenfluramina richiede uno stretto monitoraggio cardiologico con l’esecuzione di un ecocardiogramma prima dell’introduzione del farmaco, a cadenza semestrale nei primi due anni di trattamento, e annuale negli anni successivi.
Lo Stiripentolo: è il primo farmaco approvato specificatamente per la sindrome di Dravet, ed è indicato per il trattamento delle crisi tonico-cloniche, in combinazione a clobazam e a valproato. Ha un meccanismo d’azione complesso, che comprende meccanismi GABAergici multipli, inibizione dei canali del calcio e della lattato deidrogenasi3. Inoltre, incrementa la concentrazione plasmatica del clobazam; pertanto, è opportuno, in corso di titolazione dello stiripentolo, ridurre la posologia del clobazam per evitare la comparsa di sonnolenza, neutropenia asintomatica e ipotonia.
Il cannabidiolo è indicato come terapia aggiuntiva, in associazione con il clobazam, per le crisi epilettiche associate a SD, a partire dai due anni di età.
Il topiramato (Topamax), soprattutto in pazienti con fenotipo relativamente lieve e a partire dai due anni di età, può essere impiegato come terapia aggiuntiva nel caso di fallimento dei farmaci di prima, seconda e terza linea.
La dieta chetogenica e altri strumenti, come la stimolazione del nervo vago, dovrebbero essere considerati in caso fallimento dei farmaci consigliati, in aggiunta alla terapia farmacologica.
Crisi prolungate e protocollo di intervento
Molte persone con SD possono essere soggette a crisi epilettiche prolungate che necessitano di un intervento di emergenza, eventualmente anche al domicilio.
Le famiglie dovrebbero perciò disporre del farmaco di soccorso da somministrare a domicilio secondo indicazione medica nel caso di crisi prolungata. I farmaci di soccorso generalmente utilizzati sono il diazepam per via rettale e il midazolam (Buccolam) per via buccale.
Al fine di somministrare al paziente in modo tempestivo il trattamento più appropriato per l’interruzione della crisi, secondo la sua storia clinica, è importante che il genitore richieda allo specialista un piano di intervento personalizzato che riporti tutte le indicazioni utili per il trattamento in emergenza sia al domicilio, sia in ambiente ospedaliero.
Sperimentazione clinica
Nell’ultimo decennio la sindrome di Dravet (SD) ha ricevuto un’attenzione sempre crescente da parte dell’industria farmaceutica. Attualmente, accanto ai farmaci già approvati e disponibili, a livello internazionale sono in fase di sperimentazione clinica numerose opzioni terapeutiche, che si differenziano tra loro sia per lo stadio di sviluppo in cui si trovano che per il loro meccanismo d’azione. E’ un dato di fatto che, il panorama terapeutico della SD si sta spostando dai più tradizionali trattamenti sintomatici, che agiscono cioè sui sintomi della malattia (in particolare sulle crisi epilettiche), ai trattamenti disease-modifying cioè che mirano ad intervenire sulle cause stesse della malattia, agendo quindi non solo sul disturbo epilettico ma anche sui disturbi del neurosviluppo, dei sintomi comportamentali e della disabilità motoria.
Le sperimentazioni cliniche attualmente attive in Italia, con ingaggio dei pazienti in corso, sono le seguenti:
1) Studio in aperto, braccio unico, per valutare la sicurezza, la tollerabilità e la farmacocinetica della Fenfluramina (Hydrochloride) in bambini con Sindrome di Dravet da 1 a 2 anni di età (ORCHID) ClinicalTrials.gov ID NCT06118255
Centri clinici coinvolti:
Istituto Giannina Gaslini, Genova
Azienda Ospedaliera Universitaria Meyer IRCCS, Firenze
Policlinico Universitario Fondazione Agostino Gemelli IRCCS, Roma
Ospedale pediatrico Bambino Gesù, Roma
Pipeline di sperimentazione clinica per la SD a livello internazionale.
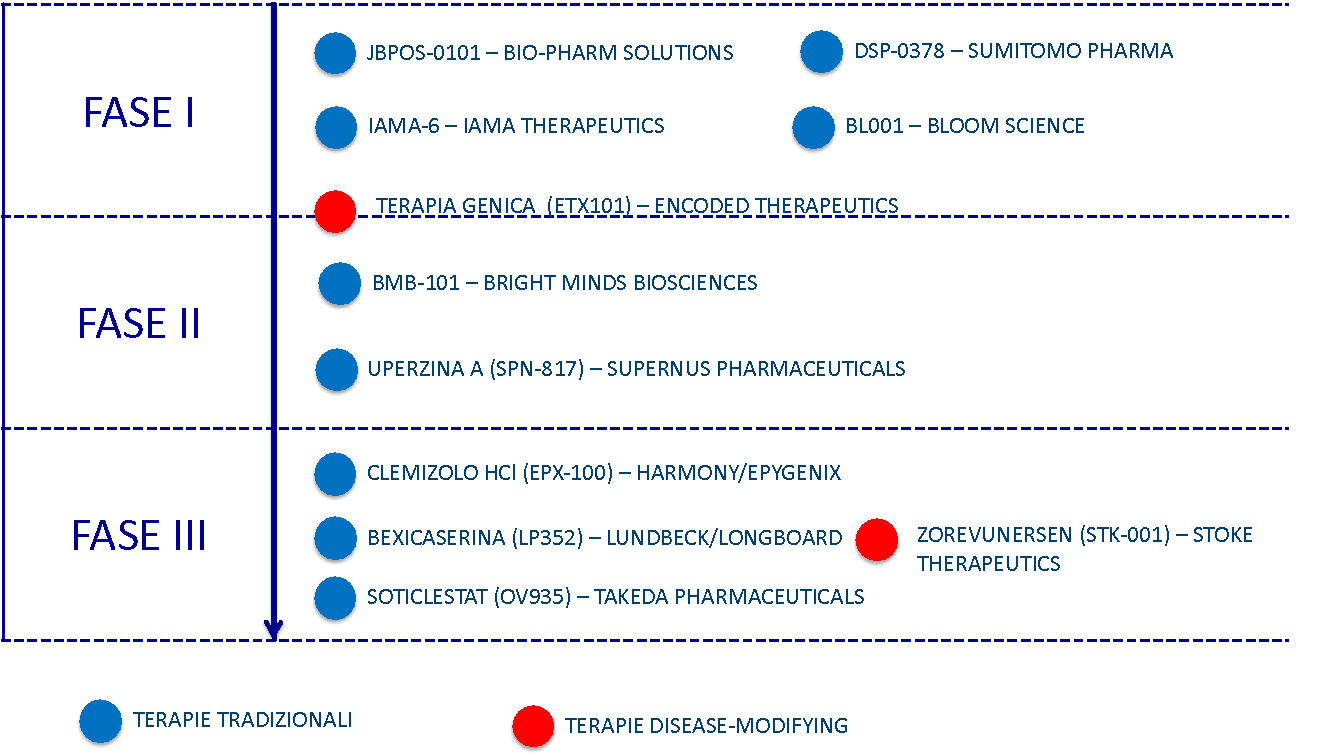
In totale, si registrano attualmente 10 programmi nelle tre fasi di sviluppo clinico, di cui 8 terapie tradizionali e 2 disease-modifying.
Nella tabella seguente sono riepilogate le principali caratteristiche delle diverse strategie terapeutiche (Ana Mingorance, Dracaena Report. Dravet syndrome pipeline 2024; )..
Alcune di esse sono già state autorizzate anche in Italia ma non è ancora stata aperta la fase di ingaggio dei pazienti.
Aggiornamento terapie disponibili e in corso di sperimentazione – Dott.ssa Domenica Battaglia,Policlinico Universitario Fondazione Agostino Gemelli IRCCS, Roma – settmbre 2024
Le terapie geniche per la sindrome di Dravet – Dott.ssa Gaia Colasante, Ospedale San Raffaele, Milano – settembre 2024
APPROFONDIMENTI SULLE TERAPIE IN SPERIMENTAZIONE CLINICA

Terapie tradizionali
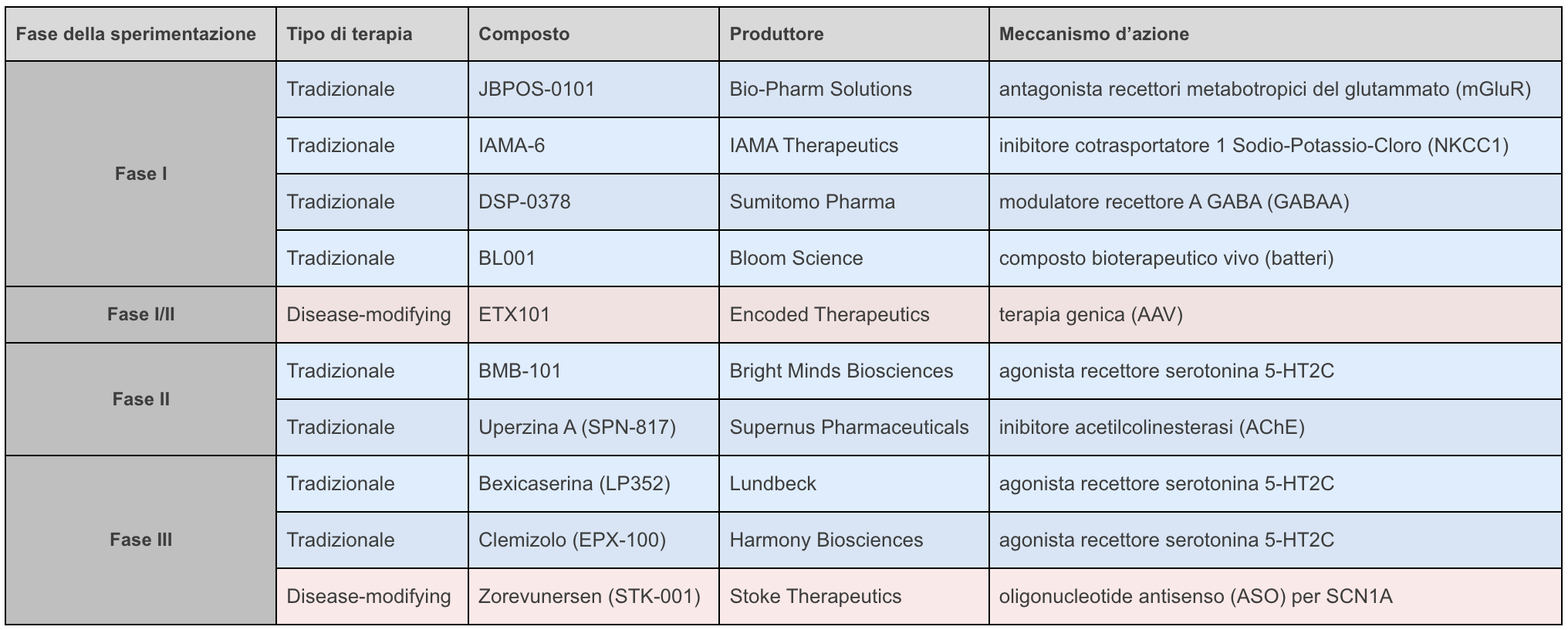
- JBPOS-0101 (Bio-Pharm Solutions): antagonista dei recettori metabotropici del glutammato, in fase di sviluppo per il trattamento dell’Alzheimer e di molteplici indicazioni per l’epilessia. Il sito web di Bio-Pharm riporta che il programma si sta “preparando per la fase 2” per la SD (http://b-psol.com/pipeline/)
- IAMA-6 (IAMA Therapeutics): inibitore del cotrasportatore 1 Sodio-Potassio-Cloruro (NKCC1), in fase di sviluppo da IAMA Therapeutics, azienda italiana con sede a Genova, per i disturbi dello spettro autistico e l’epilessia farmacoresistente. Attraverso la modulazione di NKCC1, IAMA-6 è in grado di modulare la sensibilità neuronale all’acido gamma-amminobutirrico (GABA), uno dei principali neurotrasmettitori inibitori del nostro sistema nervoso centrale. IAMA-6 è attualmente in fase di valutazione in uno studio di Fase 1 su volontari sani (NCT06300398) (https://iamatherapeutics.com/iama-therapeutics-to-present-new-preclinical-data-at-upcoming-american-epilepsy-society-2023-annual-meeting/)
- DSP-0378 (Sumitomo Pharma): composto che appartiene ad una nuova generazione di farmaci GABAergici con effetto anticrisi. È un modulatore del recettore A del GABA che agisce su vari sottotipi di recettori A del GABA in modo diverso dai modulatori GABA più tradizionali, come le benzodiazepine e i neurosteroidi (https://www.sumitomo-pharma.com/ir/library/factbook/assets/pdf/fb202311.pdf)
- BL001 (Bloom Science): prodotto bioterapeutico vivo somministrabile per via orale, composto da una combinazione proprietaria di ceppi batterici che Bloom Science sta sviluppando per il potenziale trattamento delle crisi epilettiche associate alla SD e per altre indicazioni. BL-001 è stato progettato in seguito alla scoperta che la dieta chetogenica induce cambiamenti specifici nel microbioma intestinale e che questi particolari cambiamenti potrebbero essere necessari per l’attività anticrisi della dieta chetogenica. BL-001 mira a ricreare questo beneficio terapeutico somministrando le popolazioni batteriche chiave che portano a cambiamenti nell’ipereccitabilità cerebrale, in parte modulando il sistema GABAergico. Nel 2023, BL-001 ha ricevuto dall’FDA l’Orphan Drug Designation1.
- (farmaco o prodotto biologico studiato per prevenire, diagnosticare o trattare una malattia o una condizione rara) per la SD. BL-001 è stato valutato in uno studio di Fase I su volontari sani, negli Stati Uniti , completato nel 2023 (NCT05818306). Bloom intende portare questo programma a studi di Fase II con indicazioni mirate per la SD e altre encefalopatie epilettiche e dello sviluppo. Il sito web dell’azienda indica BL-001 come pronto per la Fase II per la SD, anche se al momento l’azienda non ha ancora fatto alcun annuncio sull’avvio della sperimentazione (https://bloomscience.com/science/)
- BMB101 (Bright Minds Biosciences): agonista della serotonina che agisce esclusivamente attraverso la via di segnalazione della proteina Gq ed evita l’attivazione della proteina beta-arrestina. Ciò è fondamentale per ridurre al minimo il rischio di desensibilizzazione del recettore e di sviluppo di tolleranza. Si tratta perciò di un farmaco anticrisi dal meccanismo innovativo, progettato per fornire un sollievo prolungato dalle crisi in popolazioni di pazienti affetti da epilessia farmacoresistente. Nel 2023, l’azienda ha completato uno studio di Fase I su volontari sani (NCT05397041), e nel 2024 ha annunciato l’avvio di uno studio di Fase II in Australia per arruolare 20 pazienti con epilessia da assenza e con encefalopatie epilettiche dello sviluppo “come la SD e la sindrome di Lennox-Gasteau” di età compresa tra i 18 e i 65 anni (NCT06401538). Secondo la casa produttrice, il farmaco potrebbe anche avere effetti positivi nel parziale recupero della disabilità cognitiva (https://brightmindsbio.com/pipeline/)
- Uperzina A – SPN-817 (Supernus Pharmaceuticals): inibitore dell’acetilcolinesterasi originariamente estratto da una pianta utilizzata nella medicina tradizionale cinese. Nel 2017 SPN-817 ha ricevuto dall’FDA l’Orphan Drug Designation2per la SD. Sebbene nel comunicato stampa di acquisizione Supernus abbia menzionato l’intenzione di sviluppare SPN-817 per il trattamento della SD (comunicato stampa del settembre 2018), Supernus non ha rilasciato alcuna indicazione sulla SD come potenziale indicazione per questo programma in seguito. Dopo aver completato gli studi di Fase I per la nuova formulazione, Supernus ha avviato uno studio di Fase II in pazienti adulti con crisi epilettiche resistenti al trattamento (NCT05518578) e nel maggio 2024 ha annunciato di essere riusciti ad ottenere una significativa riduzione delle crisi nei pazienti trattati (https://www.supernus.com/research-development)
- Bexicaserina – LP352 (Lundbeck): agonista selettivo del recettore della serotonina 5-HT2C. Nel 2024, la bexicaserina ha ottenuto dall’FDA l’Orphan Drug Designation (farmaco o prodotto biologico studiato per prevenire, diagnosticare o trattare una malattia o una condizione rara) per il trattamento delle crisi epilettiche nella SD. A differenza della fenfluramina, la bexicaserina non richiede un monitoraggio ecocardiografico durante l’assunzione, in quanto non ha effetto sul recettore responsabile degli effetti collaterali cardiaci e polmonari della fenfluramina. Longboard ha dapprima condotto uno studio di Fase II (studio PACIFIC, NCT05364021) su 52 pazienti con encefalopatie epilettiche dello sviluppo di età compresa tra 12 e 65 anni. In questo studio erano stati arruolati solo 4 pazienti con SD, di cui solo 3 lo hanno portato a compimento, mostrando un’efficacia paragonabile a quella della fenfluramina. Nel 2024, Longboard ha annunciato la pianificazione di uno studio Fase III per sindrome di Dravet (studio DEEp SEA, NCT06660394) e di uno studio aperto a diverse encefalopatie epilettiche dello sviluppo (DEEp OCEAN, NCT06719141). Lo studio di fase 3 sulla SD, avviato a settembre 2024, mira a reclutare circa 160 pazienti di età compresa tra i 2 e i 65 anni in 80 siti a livello globale (https://www.lundbeck.com/global/our-science/pipeline)
- Clemizolo Cloridrato – EPX-100 (Harmony Biosciences): antistaminico che agisce attraverso la modulazione della serotonina. In passato è stato utilizzato per il trattamento delle allergie, e studi preclinici in modello animale di zebrafish hanno dimostrato che EPX-100 può potenzialmente contribuire a ridurre le crisi epilettiche nei pazienti con SD. Nel settembre 2020, Epygenix (ora acquisita da Harmony Biosciences) ha avviato uno studio di Fase II (studio ARGUS, NCT04462770) su 24 pazienti. Ora lo studio è stato ampliato in Fase III per reclutare fino a 100 partecipanti di età superiore a due anni. Lo studio è attualmente in fase di reclutamento negli Stati Uniti, in Canada, Asia, ed Europa (in particolare in Polonia, Ungheria e Spagna) (https://argustrial.com/?_sft_region=united-states).
- Soticlestat – OV935 (Takeda Pharmaceuticals): inibitore altamente selettivo dell’enzima colesterolo 24 idrossilasi, sviluppato congiuntamente da Takeda e Ovid Therapeutics per il trattamento di rare sindromi epilettiche e dello sviluppo, tra cui la SD. Grazie all’inibizione dell’enzima colesterolo 24 idrossilasi, il farmaco riduce la segnalazione glutammatergica e la neuroinfiammazione, con un impatto positivo sulla suscettibilità alle crisi epilettiche. Nel 2017 ha ottenuto dall’FDA l’Orphan Drug Designation (farmaco o prodotto biologico studiato per prevenire, diagnosticare o trattare una malattia o una condizione rara) per il trattamento delle crisi epilettiche nella SD. Il farmaco è stato testato in uno studio clinico di Fase III per la sindrome di Dravet (SKYLINE, NCT04940624) in 144 pazienti di età compresa tra i 2 e i 21 anni. I risultati ottenuti hanno mostrato una riduzione mensile del 22% nella frequenza delle crisi convulsive per i pazienti trattati con soticlestat rispetto all’8,6% con placebo, il trial non ha potuto dimostrare l’endpoint primario. In data 30 gennaio 2025 Takeda ha annunciato la decisione di interrompere il programma di sviluppo di soticlestat (TAK-935).
Terapie disease-modifying
- ETX101 (Encoded Therapeutics): approccio di terapia genica basato su vettori adenovirali (AAV) per aumentare la trascrizione del gene SCN1A e di ripristinare l’aploinsufficienza3
(condizione in cui viene a mancare una delle due copie di geni presenti nelle cellule) nei pazienti con SD causata da mutazioni del gene SCN1A. L’approccio proposto è specificatamente progettato per agire, con un’unica somministrazione, sulla causa alla base della patologia, trattando potenzialmente l’intera gamma di manifestazioni convulsive, cognitive, comportamentali, motorie e di sviluppo associate alla SD. ETX101 ha ottenuto dall’FDA l’Orphan Drug Designation (farmaco o prodotto biologico studiato per prevenire, diagnosticare o trattare una malattia o una condizione rara) per il trattamento della sindrome di Dravet associata a mutazioni del gene SCN1A nel 2020. SCN1A è stato per lungo tempo ritenuto non idoneo alla terapia genica basata su AAV a causa delle sue grandi dimensioni, ma Encoded è stata in grado di superare tali limitazioni esprimendo un fattore di trascrizione ingegnerizzato per aumentare la regolazione di SCN1A specificamente nelle cellule GABAergiche. Nel 2024, Encoded ha avviato un programma clinico di Fase I/II per ETX101, denominato POLARIS, composto da tre studi clinici distinti: ENDEAVOR (NCT05419492), WAYFINDER (NCT06112275), e EXPEDITION (NCT06283212). Durante il congresso annuale 2024 dell’American Epilepsy Society (dicembre 2024), Encoded ha annunciato che 5 pazienti con SD hanno già ricevuto la terapia, somministrata per via intracerebroventricolare. Lo studio ENDEAVOR sta reclutando, negli Stati Uniti, bambini di età compresa tra i 6 mesi a 3 anni in una Fase I in cui verranno valutate fino a tre dosi di ETX101 in “add-on”, quindi i pazienti devono assumere almeno un farmaco anticrisi già sul mercato. In una Fase II successiva, lo studio prevede di arruolare fino a 18 partecipanti. Lo studio WAYFINDER è localizzato in Australia e sta reclutando pazienti di età compresa tra i 3 e i 7 anni, valutando fino a tre dosi di terapia. Anche questo studio viene condotto come “add-on”. Infine, lo studio EXPEDITION sta reclutando nel Regno Unito bambini di età compresa tra i 6 mesi e 4 anni, testando fino a tre dosi di ETX101 in “add-on”. I risultati saranno disponibili nel corso del 2025 (https://encoded.com/programs/etx101-for-dravet-syndrome/). - Zorevunersen – STK-001 (Stoke Therapeutics): è stato il primo trattamento disease-modifying per la SD per cui è stata avviata la sperimentazione clinica. Si tratta di una terapia a base di oligonucleotidi antisenso4(ASO) somministrata per via intratecale nel liquido spinale. STK-001 si basa su un approccio chiamato TANGO (Targeted Aumentation of Nuclear Gene Output), e prevede appunto l’utilizzo di un ASO che si lega alle molecole di pre-RNA messaggero per aiutare i geni funzionanti a essere tradotti, ripristinando così la produzione della proteina “sana” mancante. Nel 2019 ha ottenuto dall’FDA l’Orphan Drug Designation5 per il trattamento della SD associata a mutazioni del gene SCN1A. Zorevunersen è stato testato in pazienti con SD in due studi paralleli di Fase I/II: MONARCH (NCT04442295) negli Stati Uniti, e ADMIRAL (ISRCTN99651026) nel Regno Unito. Nello studio MONARCH sono stati arruolati 62 pazienti di età compresa tra 2 e 18 anni con mutazione nel gene SCN1A, mentre nello studio ADMIRAL la terapia è stata testata in 19 bambini e adolescenti con SD. Nel dicembre 2024, al Congresso annuale dell’American Epilepsy Society, Stoke Therapeutics ha reso nota un’analisi dettagliata dei risultati dei due studi, che ha permesso di osservare una riduzione delle crisi epilettiche del 75 – 85%, che può arrivare fino all’87% all’ottavo mese di trattamento, a fronte di una buona tollerabilità della terapia da parte dei pazienti. Inoltre, il trattamento si è dimostrato in grado di apportare miglioramenti significativi anche per la sfera cognitiva, comportamentale e motoria, in particolare per quanto riguarda la comunicazione, le capacità grosso-motorie6, la postura, le relazioni interpersonali, l’attenzione e il linguaggio. Attualmente Stoke Therapeutics ha in programma, per il secondo quadrimestre del 2025, uno studio clinico di Fase III denominato EMPEROR, localizzato negli Stati Uniti, nel Regno Unito, in Europa e in Giappone (https://www.stoketherapeutics.com/disease-areas/dravet-syndrome/).
Nel febbraio 2025, l’azienda farmaceutica Biogen ha annunciato di aver avviato una collaborazione con Stoke Therapeutics per la commercializzazione di Zorevunersen al di fuori degli Stati Uniti (https://investors.biogen.com/news-releases/news-release-details/biogen-and-stoke-therapeutics-enter-collaboration-develop-and).
Conosci la sperimentazione clinica
La sperimentazione clinica rappresenta una componente essenziale della ricerca medica basata su evidenze e prevede il coinvolgimento di persone che, a seconda delle diverse fasi, possono essere volontari sani o pazienti.
L’obiettivo della sperimentazione clinica è quello di esaminare e ottenere prove sufficienti sulla sicurezza e l’efficacia di un nuovo trattamento (farmaco, dispositivo medico, procedura chirurgica o esame diagnostico).
La sperimentazione generalmente si svolge in 3 fasi. Per ogni fase, il disegno e la pianificazione, sono contenuti nel protocollo, che viene approvato dal Comitato Etico del centro ospedaliero presso il quale vengono svolti gli studi, nonché dal Ministero della Sanità. Nel processo autorizzativo, si tiene conto, tra l’altro, dei risultati della sperimentazione preclinica (Fase 0), che viene condotta in laboratorio su modelli cellulari e in vivo su modelli animali, per valutare l’efficacia potenziale e i potenziali rischi e tossicità per l’uomo.
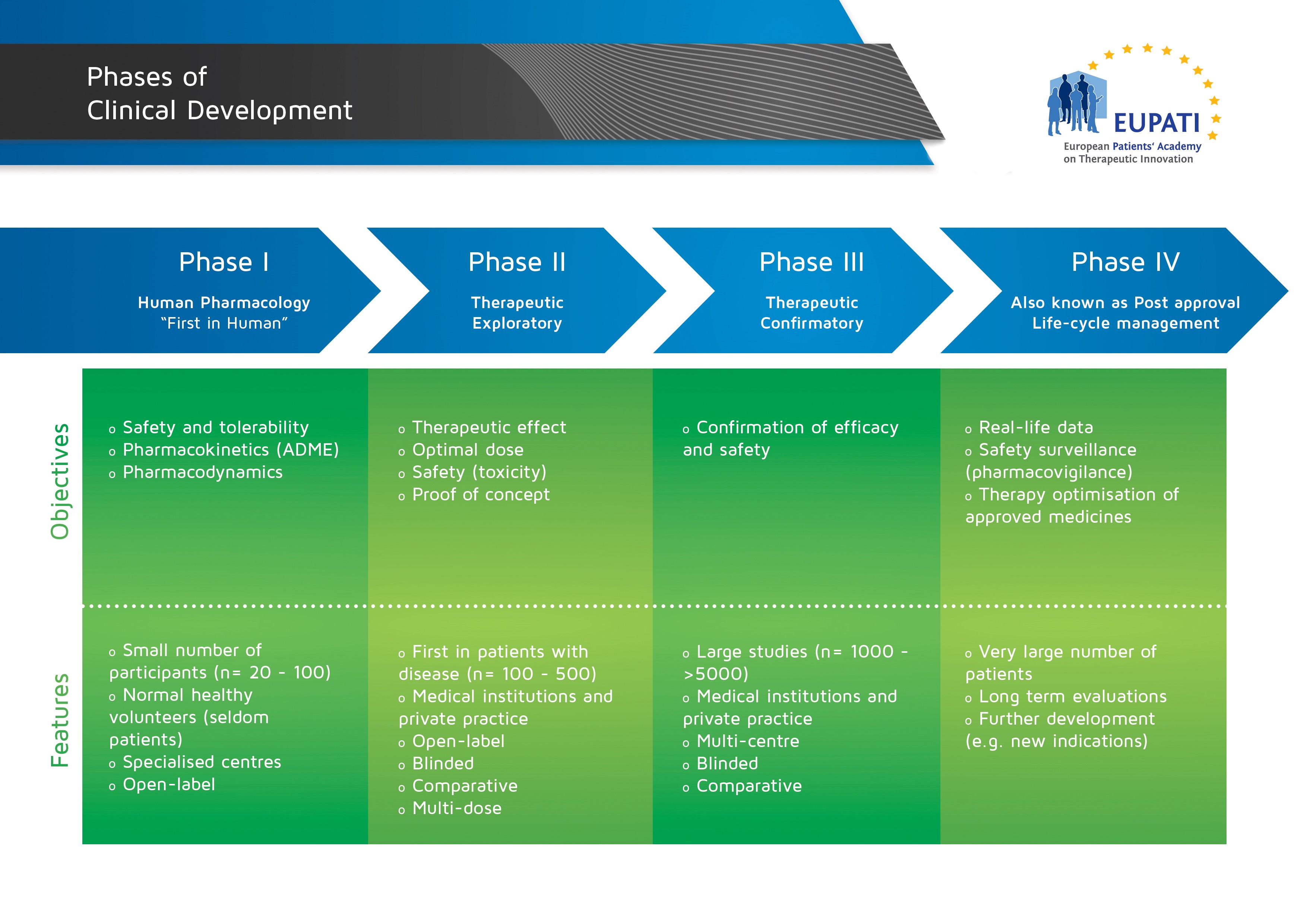
Fasi della sperimentazione clinica.
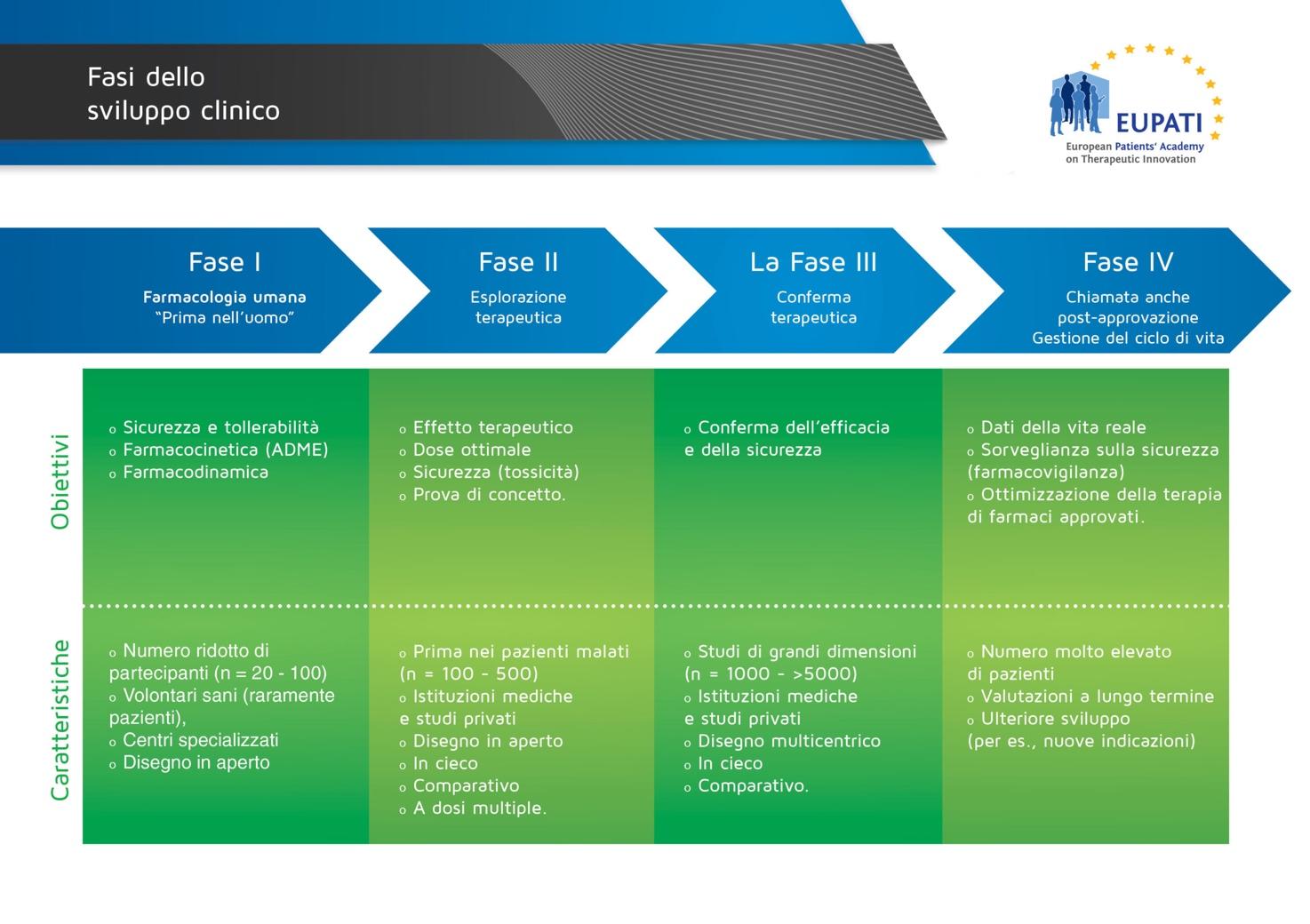
Phases of Clinical Development” by EUPATI is licensed under CC BY-NC-SA 4.0.
{kind=link}
- La fase I viene svolta generalmente su volontari sani mentre le fasi II e III vengono eseguite con persone malate seguendo diversi protocolli; il più utilizzato è quello in doppio cieco, nel quale i pazienti vengono casualmente divisi in due gruppi di trattamento (gruppo placebo, quando eticamente possibile, o gruppo farmaco in sperimentazione). Né i pazienti, né gli operatori sanitari conoscono la divisione dei gruppi.
- La Fase III è una sperimentazione che si svolge in diversi centri ospedalieri, per confermare l’efficacia della terapia, affinare dosaggi e formulazione, superare il problema della variabilità individuale, che può causare reazioni diverse in pazienti diversi, e determinare se il farmaco dimostra di avere maggiori benefici rispetto a farmaci simili già in commercio (rapporto rischio/beneficio).
In questa fase si utilizza lo studio clinico controllato randomizzato, nel quale i pazienti vengono divisi casualmente in due gruppi di trattamento (gruppo placebo, quando eticamente possibile, o gruppo farmaco in sperimentazione). L’attribuzione casuale del nuovo farmaco o del placebo/farmaco di controllo garantisce che i due gruppi siano il più possibile simili per tutte le caratteristiche salvo che per il composto assunto.
Durante questa fase vengono controllate con molta attenzione l’insorgenza, la frequenza e gravità di possibili effetti indesiderati. La durata della somministrazione del farmaco è variabile a seconda degli obiettivi che la sperimentazione si pone, ma in genere dura dei mesi.
Terminata la fase III di sperimentazione, se gli obiettivi dello studio sono stati raggiunti, la casa farmaceutica può effettuare la domanda per l’autorizzazione all’immissione in commercio. Nella maggioranza di casi la domanda viene presentata all’Agenzia Europea del Farmaco (EMA) seguendo la procedura centralizzata.
Dopo l’immissione di un farmaco sul mercato segue la Fase IV (post-marketing) per valutare, in un usuale contesto di prescrizione, il valore terapeutico e/o gli effetti collaterali (farmacovigilanza).
3Bacq A, et al. An Update on Stiripentol Mechanisms of Activation: A Narrative Review. Adv Ther 2024. doi: 10.1007/s12325-024-02813-0; doi: 10.1007/s12325-024-02813-0; https://link.springer.com/article/10.1007/s12325-024-02813-0